Yung-Che Kuo, Ph.D. 1 & Yen-Hua Huang, Ph.D. 1,2,3
1 TMU Research Center for Cell Therapy and Regeneration Medicine, Taipei Medical University, Taipei, Taiwan.
2 Department of Biochemistry and Molecular Cell Biology, School of Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan
3 International Ph.D. Program in Cell Therapy and Regenerative Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan
Abstract
Nicotine, a genotoxic agent and tumor-promoting factor, has been linked to various types of cancer. This commentary highlights recent findings demonstrating that nicotine exposure promotes tumor malignancy in triple-negative breast cancer (TNBC) through coordinated activation of the nicotinic acetylcholine receptors α9 (CHRNA9) and insulin-like growth factor-1 receptor (IGF1R) signaling axis. Evidence from multiple experimental approaches and models, including cellular systems, animal studies, patient cohorts, and public datasets, indicates that elevated expression of CHRNA9, IGF1R, and stemness-related genes correlates with poor survival in patients with TNBC. Furthermore, nicotine enhances cancer stemness properties, invasion, metastasis, and tumor progression, whereas genetic silencing of IGF1R effectively suppresses these malignant phenotypes and prolongs survival in vivo. Mechanistically, IGF1R functions as a critical downstream mediator sustaining nicotine-induced stemness programs. Collectively, these findings have important translational implications, identifying the CHRNA9-IGF1R regulatory axis as a potential target for biomarker-guided therapeutic intervention. Beyond oncology, this study also emphasizes the broader public health significance of environmental nicotine exposure, suggesting its potential impact on tumor aggressiveness and cancer survivorship.
Keywords
Nicotine; Environmental exposure; TNBCs; IGF1R; CHRNA9; Stemness; Metastasis; Recurrence; Survival
Background
Accounting for roughly 10-20% of breast cancers, triple-negative breast cancer (TNBC) is an aggressive subtype marked by a high risk of early relapse and a lack of effective targeted therapeutic strategies. Cigarette smoking has been epidemiologically linked to poorer clinical outcomes in breast cancer patients [1]. Despite nicotine being a principal constituent of tobacco smoke and e-cigarette vapor, its mechanistic contribution to TNBC progression remains largely unresolved. Our recent study in the Journal of Pathology elucidates how nicotine promotes malignancy in TNBC [2]. In this study, we define the mechanistic link between nicotine exposure and stemness-related TNBC tumor progression and demonstrate IGF1R as a promising therapeutic target to reduce tumor aggressiveness in TNBC patients (Figure 1).
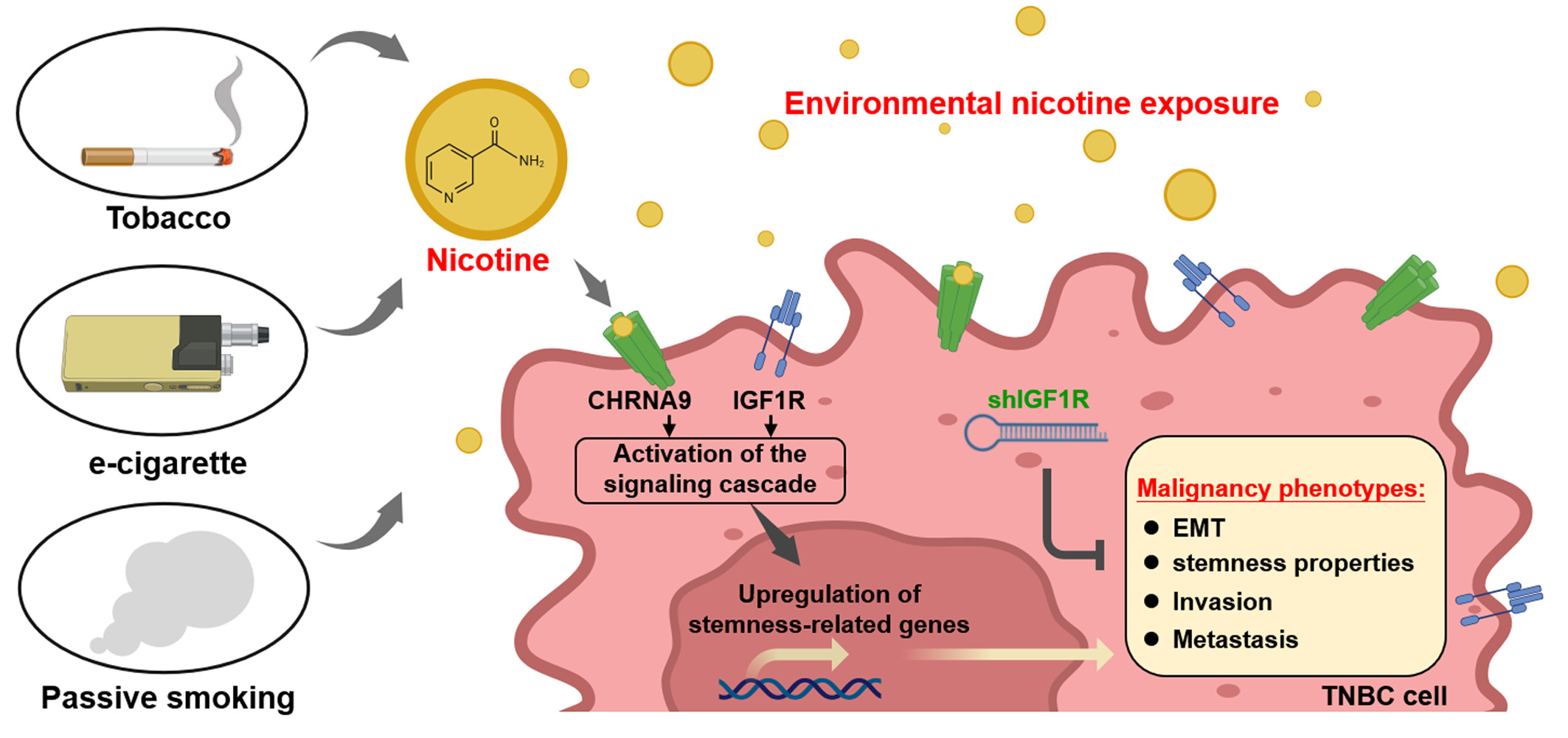
Figure 1. Brief diagram of connecting nicotine signaling with cancer stemness and metastatic progression in Triple-Negative Breast Cancer. Nicotine is a principal constituent of tobacco smoke and e-cigarette vapor; in addition, nicotine exposure also comes from passive (secondhand) smoking. Our findings reveal that nicotine significantly upregulates CHRNA9 and IGF1R, and activates signaling cascades to promote stemness-related gene expression, thereby enhancing malignancy phenotypes in TNBC cells. Moreover, knockdown of IGF1R expression diminishes nicotine/CHRNA9-induced malignancy phenotypes in vitro and in vivo. This diagram was created in BioRender. https://BioRender.com
Establish a clinically relevant role of IGF1R and CHRNA9 in TNBC
We demonstrate the clinical relevance of nicotinic acetylcholine receptors α9 (CHRNA9) and IGF1R signaling in TNBC by integrating a large public dataset (n = 299 TNBC cases) and two patient cohorts (n = 67 and n = 42). Kaplan-Meier survival analyses of TNBC cases reveal that elevated expressions of CHRNA9, IGF1R, IGF2, and stemness-related genes, including SOX2 and POU5F1, are significantly associated with poorer relapse-free survival (RFS) and distant metastasis-free survival (DMFS). Gene expression analyses of TNBC tumor tissues further show strong positive correlations between CHRNA9, IGF1R signaling components, and stemness markers, indicating coordinated activation of a stemness-related oncogenic network. Protein-level validation using tissue microarrays confirms increased expression and co-localization of CHRNA9, IGF1R/p-IGF1R, and POU5F1 in tumor tissues compared with normal breast tissues. Moreover, multivariate clinical analyses further demonstrate that high expression or co-expression of these molecules significantly correlated with tumor recurrence, supporting their role as prognostic indicators of aggressive disease behavior in TNBC [2].
Nicotine exposure enhances tumor malignancy
Using TNBC cellular and animal models, we demonstrate how nicotine exposure promotes stemness-associated malignant phenotypes in TNBC through coordinated regulation of IGF1R and CHRNA9 signaling. Nicotine exposure significantly upregulates CHRNA9 expression in TNBC cells, thereby increasing stemness-related proteins, such as POU5F1. Functional assays also demonstrate that nicotine enhances cancer stemness properties, as reflected by increased ALDH-positive cell populations, migration, and invasion abilities. Importantly, mechanistic investigations further reveal that IGF1R functions as a critical downstream regulator of nicotine-induced stemness. RNA interference targeting IGF1R markedly reduces the expression of CHRNA9, stemness markers, and epithelial-mesenchymal transition (EMT)-related genes. Knockdown of IGF1R significantly attenuates these nicotine-induced multiple malignant effects, indicating that IGF1R signaling is essential for maintaining nicotine-mediated cancer stemness programs in TNBC [2].
To further evaluate the biological relevance of highly metastatic disease, we have also established metastatic TNBC animal models. Highly metastatic cells exhibit high levels of IGF1R, CHRNA9, and stemness-associated proteins, along with enhanced invasive capacity. Notably, silencing IGF1R substantially reduces stemness properties, suppresses nicotine-induced lung metastasis, and inhibits tumor progression in vivo, ultimately prolonging survival in xenograft mouse models.
Perspectives and implications for clinical research and public health
Our findings provide important translational evidence demonstrating that nicotine exposure directly influences TNBC tumor biology rather than acting solely as a cancer-initiating factor. Nicotine exposure from tobacco smoking, e-cigarette vapor, and passive (secondhand) smoking may promote TNBC tumors with enhanced stemness characteristics, metastatic potential, and chemotherapy-drug resistance [2, 3], thereby contributing to poorer clinical outcomes observed in TNBC patients.
Clinically, our study identifies a potential biomarker-defined TNBC subgroup characterized by elevated CHRNA9 and IGF1R signaling. Previous failures of IGF1R-targeted therapies may have resulted from non-stratified clinical trials, whereas patient selection based on nicotine-associated signaling activation may improve therapeutic responses [4]. On the other hand, it also highlights opportunities to investigate nicotinic receptor antagonists or repurposed agents targeting cholinergic signaling pathways [5].
Finally, from a public health perspective, our study expands the traditional cancer prevention paradigm by suggesting that environmental nicotine exposure may influence not only cancer incidence but also tumor aggressiveness, recurrence, and patient survival. Continued nicotine exposure following cancer diagnosis may contribute to disease progression, raising concerns regarding persistent smoking and increasing e-cigarette use. These findings highlight the importance of incorporating environmental exposure history, including smoking and vaping behaviors, into oncologic risk assessment and precision prevention strategies. Moreover, populations with higher tobacco exposure may be disproportionately affected by biologically more aggressive cancers, implying a potential health risk. Collectively, the results support future cancer control approaches that integrate molecular oncology with behavioral interventions and environmental health policies, thereby bridging individual cancer care with population-level health management.
Acknowledgements
This study was supported by the National Science and Technology Council, Taiwan (NSTC 112-2320-B-038-011-MY3).
References
[1] Z. Khodabandeh, M. Valilo, K. Velaei, A. Pirpour Tazehkand, The potential role of nicotine in breast cancer initiation, development, angiogenesis, invasion, metastasis, and resistance to therapy, Breast Cancer, 29 (2022) 778-789. DOI: 10.1007/s12282-022-01369-7
[2] Y.C. Kuo, C.L. Chen, K.L. Lee, H.F. Wang, V.J. Drew, P.C. Lan, Y.S. Ho, Y.H. Huang, Nicotine-driven enhancement of tumor malignancy in triple-negative breast cancer via additive regulation of CHRNA9 and IGF1R, J Pathol, 266 (2025) 230-245. DOI: 10.1002/path.6423
[3] Y.C. Lin, Y.S. Ho, Y.C. Liao, H.S. Chang, S.H. Tu, M.H. Pan, T.C. Cheng, L.C. Chen, Inotilone suppresses fibronectin 1 expression and inhibits the growth of triple-negative breast cancer xenografts under prolonged nicotine exposure, Ecotoxicol Environ Saf, 306 (2025) 119303. DOI: 10.1016/j.ecoenv.2025.119303
[4] K.L. Lee, Y.C. Kuo, Y.S. Ho, Y.H. Huang, Triple-Negative Breast Cancer: Current Understanding and Future Therapeutic Breakthrough Targeting Cancer Stemness, Cancers (Basel), 11 (2019). DOI: 10.3390/cancers11091334
[5] C. Simpkins, E. Toska, Sparking malignancy: nicotine as a driver of stemness and metastasis in triple-negative breast cancer†, J Pathol, 267 (2025) 367-370. DOI: 10.1002/path.6475

Dr. Kuo is an assistant research fellow of TMU Research Center of Cell Therapy and Regeneration Medicine member and TMU Research Center of Thoracic Medicine.
He is also a Director of Core Laboratory of Good Tissue Practice (GTP), Taipei Medical University, Taipei, Taiwan.

Leave a comment